Авторы: Alexander A. Kolganov, Anton A. Gabrienko,, Alexander G. Stepanov ,
Abstract
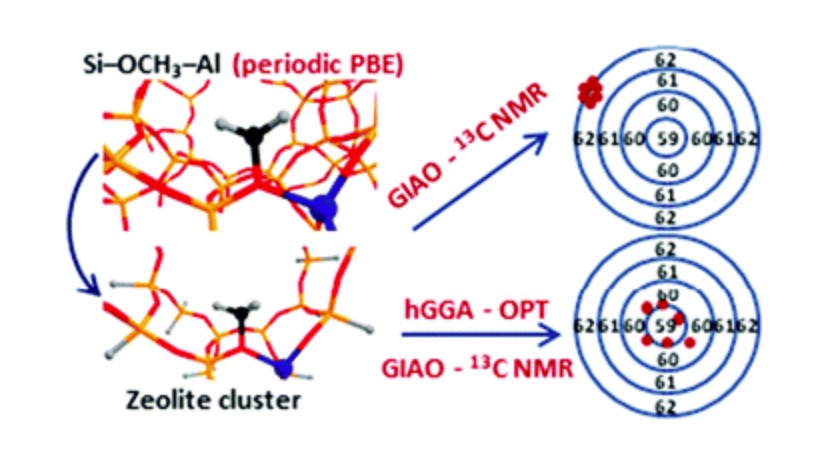
The influence of the model and method choice on the DFT predicted 13C NMR chemical shifts of zeolite surface methoxide species has been systematically analyzed. Twelve 13C NMR chemical shift calculation protocols on full periodic and hybrid periodic–cluster DFT calculations with varied structural relaxation procedures are examined. The primary assessment of the accuracy of the computational protocols has been carried out for the Si–O(CH3)–Al surface methoxide species in ZSM-5 zeolite with well-defined experimental NMR parameters (chemical shift, δ(13C) value) as a reference. Different configurations of these surface intermediates and their location inside the ZSM-5 pores are considered explicitly. The predicted δ value deviates by up to ±0.8 ppm from the experimental value of 59 ppm due to the varied confinement of the methoxide species at different zeolite sites (model accuracy). The choice of the exchange–correlation functional (method accuracy) introduces ±1.5 ppm uncertainty in the computed chemical shifts. The accuracy of the predicted 13C NMR chemical shifts for the computational assignment of spectral characteristics of zeolite intermediates has been further analyzed by considering the potential intermediate species formed upon methane activation by Cu/ZSM-5 zeolite. The presence of Cu species in the vicinity of surface methoxide increases the prediction uncertainty to ±2.5 ppm. The full geometry relaxation of the local environment of an active site at an appropriate level of theory is critical to ensure a good agreement between the experimental and computed NMR data. Chemical shifts (δ) calculated via full geometry relaxation of a cluster model of a relevant portion of the zeolite lattice site are in the best agreement with the experimental values. Our analysis indicates that the full geometry optimization of a cluster model at the PBE0-D3/6-311G(d,p) level of theory followed by GIAO/PBE0-D3/aug-cc-pVDZ calculations is the most suitable approach for the calculation of 13C chemical shifts of zeolite surface intermediates.