Authors: Nikolai Shirokii, Yevgeniya Din, Ilya Petrov, Yurii Seregin, Sofia Sirotenko, Razlivina J.S., Serov N.S., Vinogradov V.V.
Small, 2023, DOI:10.1002/smll.202207106
Abstract
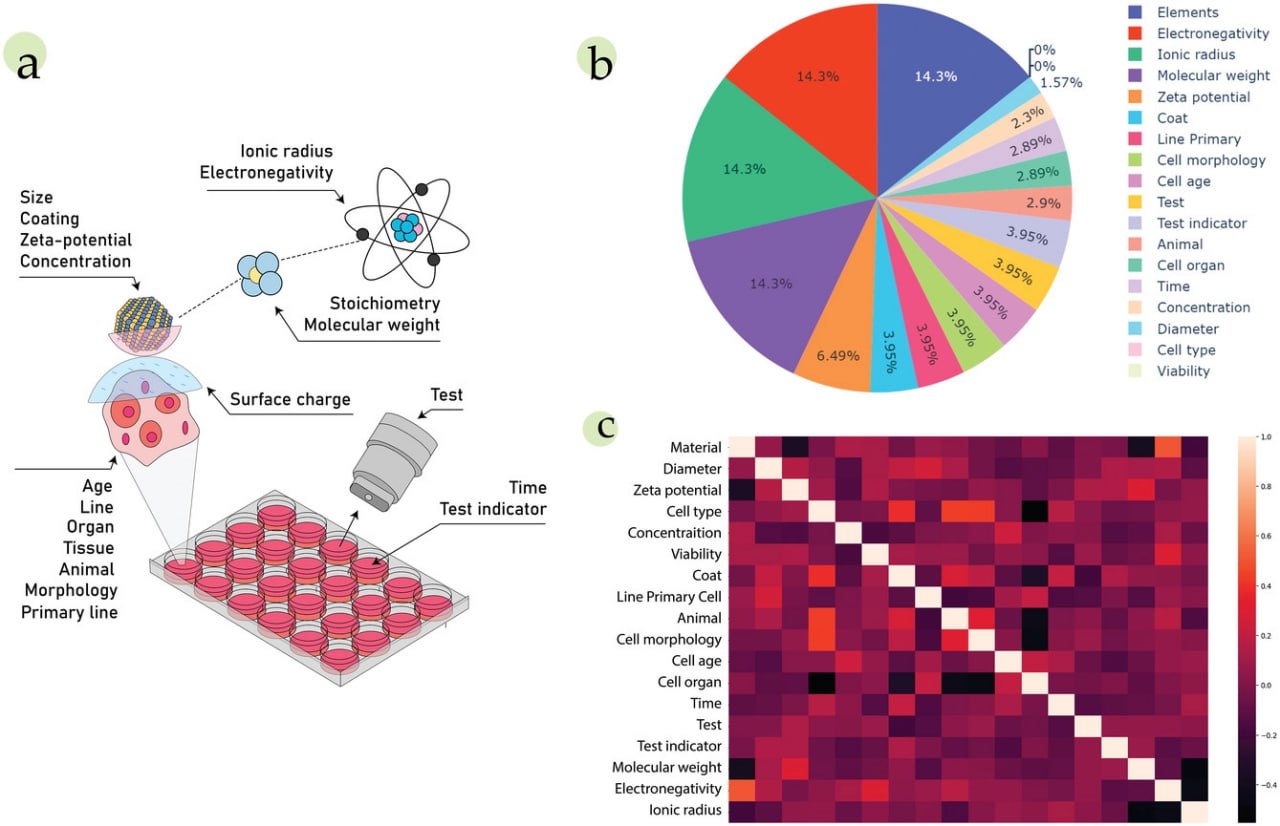
Organic chemistry has seen colossal progress due to machine learning (ML). However, the translation of artificial intelligence (AI) into materials science is challenging, where biological behavior prediction becomes even more complicated. Nanotoxicity is a critical parameter that describes their interaction with the living organisms screened in every bio-related research. To prevent excessive experiments, such properties have to be pre-evaluated. Several existing ML models partially fulfill the gap by predicting whether a nanomaterial is toxic or not. Yet, this binary categorization neglects the concentration dependencies crucial for experimental scientists. Here, an ML-based approach is proposed to the quantitative prediction of inorganic nanomaterial cytotoxicity achieving the precision expressed by 10-fold cross-validation (CV) Q2 = 0.86 with the root mean squared error (RMSE) of 12.2% obtained by the correlation-based feature selection and grid search-based model hyperparameters optimization. To provide further model flexibility, quantitative atom property-based nanomaterial descriptors are introduced allowing the model to extrapolate on unseen samples. Feature importance is calculated to find an interpretable model with optimal decision-making. These findings allow experimental scientists to perform primary in silico candidate screening and minimize the number of excessive, labor-intensive experiments enabling the rapid development of nanomaterials for medicinal purposes.