It was not so long ago that bioinformatics specialist Alexey Komissarov joined the team of ITMO University’s SCAMT Laboratory, where he conducts the assembling and annotation of complex genomes of animals and humans. At the end of 2019, the Applied Genomics research group, which he heads, received a grant from the Russian Foundation for Basic Research. Their project will be dedicated to the analysis of genes and proteins in nerve cells of lampreys, which could help in the fight against neurodegenerative diseases.

Today, you are known for your expertise in the field of bioinformatics, but your PhD thesis, which you defended in 2012, focused on the field of molecular biology. Why did you decide on bioinformatics?
Indeed, I defended my PhD thesis “Organization of large tandem repeats in the mouse genome” in the field of molecular biology under the scientific advisorship of Olga Podgornaya in 2012, but it so happened that in actuality, it was almost entirely dedicated to bioinformatics. The thing is that by that point, most issues in genome analysis could no longer have done without bioinformatics – if you can code, many tasks that could’ve taken weeks or even months to accomplish take a matter of hours. You just program an algorithm and repeat it on new data. The Institute of Cytology had a great community of self-taught bioinformatics specialists, and all of us there were slowly but surely mastering programming and genome data analysis skills.
The topic I defended my thesis on was new back then, no one had particularly explored it before. We studied the part of the genome that is virtually impossible to assemble, these are centromeres, telomeres. In other words, we have these DNA pieces that don’t have a place in the decoding of the genome, but when we analyze chromosomes with experimental methods we see that they are there, though it isn’t clear where exactly. I started to wonder about what was in that part of the genome that we couldn’t assemble, as we didn’t know exactly where, in which chromosome or in which part of the chromosome these pieces were located. When assembling the genome, such pieces are placed in an artificial chromosome – Chromosome Unknown.
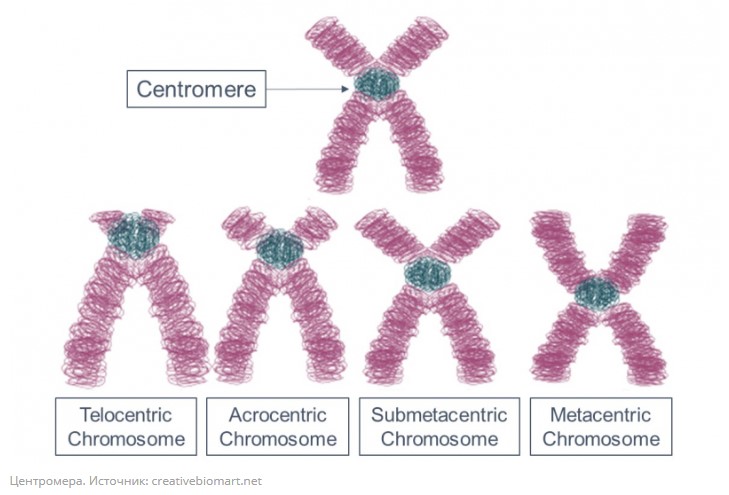
For this purpose, I wrote a program that would analyze these fragments, calling it CHRUNTA (ChrUn, from Chromosome Unknown, Tandem Analyser) – a kind of word play I couldn’t help myself including, it’s always a temptation to play with different readings in different languages, for Russians to get the joke and others to think that this is a normal name. Using this software, we found a lot of tandem repeats in the mouse genome that hadn’t been known before. However, not all answers have been obtained – for example, we still don’t know what these DNA fragments are responsible for.
And this data is needed, for example, for the creation of an artificial chromosome – such experiments are being carried out in Japan and the US. This opens previously unthinkable horizons, but it hasn’t been managed to make it stable yet, namely because there isn’t enough data on the composition of such key areas as centromeres.
Why did you make the decision to join ITMO University?
After defending my thesis, I stayed to work at the Institute of Cytology for some time, then, in 2013, St. Petersburg got two megagrants, one of them led by Stephen O’Brien. From 2013 to 2018, I worked under his guidance.
I was brought here, to ITMO University, by my acquaintances in 2019. They’d told me about the SCAMT Laboratory, how there were a lot of opportunities, young staff, lots of students, lots of interesting stuff going on. The lab needed a person for genome data analysis of cardiovascular diseases patients. This really is one of the most lively places in St. Petersburg: everybody is doing something all the time.
Apart from this, I’ve always liked interdisciplinary research; I’ve had lots of branching out in my career: I’ve worked in AI, machine learning, text analysis, as a programmer in general. When I first came here, collaborations immediately started to map out with different groups of chemists.
I was given the lead over eight Master’s students, whom we are now teaching bioinformatics. Parallel to that, we’re working on different projects: both those I’d started before coming to ITMO and the ones that started here.

Recently, one of these projects was awarded a grant from the Russian Foundation for Basic Research. Could you tell us more about this project?
We got the message that we won the grant on December 30, 2019. The project focuses on the genomics of the Mustelidae. These are very cool animals, the ultimate predators. You can find some videos on YouTube where an ermine is playing with a cat and the cat doesn’t even have enough reactive power to follow the former’s movements.
The Mustelidae are interesting because their genomes practically haven’t been sequenced. The Felidae, for example, are very well-studied, you could say that the research of this group has been divided between the scientists of the world, it’s rather difficult to discover something new here. But there are some blind spots remaining in the research of the Mustelidae group.
We’ll focus on such types as sable, yellow-throated marten, stone marten, Siberian weasel, and Vietnamese weasel. These samples will be relatively easy for us to get. In the first year, we’ll conduct a study of yellow-throated martens, animals with a yellow-green coloring. This species is part of regional red lists of threatened species in many places in Russia. We’re collaborating with Alexey Abramov from the Institute of Zoology of the Russian Academy of Sciences and a group of cytogenetics specialists led by Alexander Grafodatsky from the Institute of Molecular and Cellular Biology in Novosibirsk. Alexey Abramov goes on expeditions, he’s an expert on the Mustelidae, and as such will be able to collect samples. In their turn, our colleagues from Novosibirsk are the best in the world in making karyotypes. It was this, by the way, that has become the advantage of our grant application.

How will the research be carried out?
We get the DNA, sequence it, assemble the genome and annotate it. What does this mean? There’s a good analogy – take several editions of the complete works of Lenin so that there’d be several copies of each volume. Now cut each page into pieces of about a hundred letters, shuffle them carefully and remember that some of the printing ink has been removed. Then you’ll need to reassemble the entire edition back from these pieces and analyze what is written there. This is what we are doing.
Sequencing is the first stage, this is akin to cutting these fragments and reading them, letter by letter. We can’t read the genome completely straightaway, it’s for this that we need to the small fragments. Today, the best results are obtained by reading small pieces of 150 to 300 nucleotides. In total, the genome of the mustelid contains somewhere between 2.3 and 2.4 billion nucleotides. This is why bioinformatics plays such a role – manually, such a volume simply cannot be assembled. When all this was just at its onset, when the bacterial genomes were the ones being assembled, this could be done in Word or in Excel in a month. But the mustelid genome requires very tricky algorithms and assembling programs.
There are such methods as PacBio and Oxford Nanopore that allow you to read larger fragments. The former is much more expensive but gives a more high-quality result, the latter is cheaper and the readings are much longer, but return more errors. Both methods involve a large number of errors, though, and you have to run through the DNA many times in order to get rid of these errors, and it’s because of this such studies are very expensive.
Another method of DNA fragment readings is the link-reads method. Under it, we don’t read the whole molecule in one go – only a small part of fragments of each DNA molecule – but we know for sure from which molecule each fragment was taken. And this is the hack that allows for a much better genome sequencing. Our work will feature all three methods.
The second stage is the assembly: when we have selected our fragments and read them, they need to be assembled back into their respective volumes. Today, though, we’re capable of not only assembling them not into complete “books”, that is, chromosomes, but also, in the best-case scenarios, into separate chapters, that is, parts of chromosomes. Even the best methods aren’t perfect, however, which is why even the best assemblies resemble Swiss cheese – there are many gaps the composition of which leaves much to guessing. The last step is annotation: we try to find protein-coding genes, repeats, regulatory elements.
Currently, the main thing in working with animal genomics is to catch the animal and correctly extract the DNA. All consecutive stages depend on the correct procedure of the extraction process. If a mistake occurs during the collection of samples, all the following stages I spoke of earlier will only amass the number of errors.
What do you want to find out during this research?
For example, using this data we can uncover the structure of a population and restore its demographic history – what its size was before, how it changed. For example, sables in Siberia had been almost fully destroyed by hunters by the middle of the 20th century. Then, in the 1960s, the animals were reintroduced into the forests and the population quickly multiplied. Now it’s important to find out what this did to their genome. This is a classic bottleneck situation – which means that we’d had a large variety of genes, then the population underwent a sharp decrease, and not all DNA variations remained. Then the population saw a sharp rise in numbers, and it is interesting to see what trace this development left on it.
Speaking about more fundamental tasks, it’s interesting to identify exactly what genes make the mustelids predators. As I’ve already mentioned, these are very fast, dexterous animals that are much more dangerous than their feline counterparts under the same conditions – the ultimate predators. Some species in this group have a delay in implantation, when fertilization is separated in time from the further development of the embryo – there’s this phenomenon, but it’s not known how it occurs at the genome level. We will attempt to shed light on this.
Another remaining mystery is speciation, namely why different species have a different number of chromosomes. There are many mysteries that are going on 40 years old now, and no one’s able to find an answer.

What projects are you currently working on apart from this one?
There’s a very interesting project on the lamprey genome that’s just started. We work on it together with the Laboratory of Synapse Biology (Institute of Translational Biomedicine of St. Petersburg State University). Lamprey in St. Petersburg is almost like smelt, our local species, the market’s teeming with them when they’re in season. But it’s also a very important animal species in terms of physiology studies.
The thing is that it has very large neurons, and based on their example it’s very convenient to study the structure of neurons, their general operation. However, a number of studies encounter the limitation related to the lack of river lamprey genome. Now, a method has been developed that allows you to see what is transcribed in each specific cell – this is the single-cell RNA-seq method. This will provide revolutionary data on the functioning of the nervous system as a whole and its regeneration in particular. But in order to conduct this analysis for lampreys, we need this species’ genome.
These results will help us in researching various diseases of the nervous system – from Alzheimer’s disease to mechanical spinal injuries. The fact is that people have a lot of conserved genes common to those of different animals. When we find such conserved connections, we can use animal material for a variety of model experiments so as not to waste rare material obtained from humans. Animals give out good clues: for example, cats are an ideal model for studying the human immune system, as they and us have a lot in common in this regard.
Apart from that, under closer inspection, it transpired that lampreys have a very complex genome. And this is my main research interest: the genomes that have something wrong with them, roughly speaking; that are difficult to assemble. For instance, lampreys’ reproductive and somatic cells have a different genome size. We don’t exactly understand how this happens.
We also work with invertebrate animals. This is truly a world of miracles – whenever direction you look into, everything is interesting there. Invertebrate genomes are a challenge, you don’t know how to approach them. Generally, there are now many areas in genomic bioinformatics where it’s easy to encounter an unchartered territory. This is the thrill of science.
For example, now we have the clam genome, of the white clams that can be found literally on any market in Europe. Their genome size also varies, and we don’t understand why. They also have maternal and paternal mitochondrial DNA. In humans, they are usually transmitted only through the maternal line, but both the paternal and the maternal lines are involved. We also want to start bioinformatic work with mosquitoes that transmit various viral diseases. I wrote an article about them, and now I want to return to this topic.
There are many viruses transmitted by mosquitoes – for example, Zika and Dengue. Now that’s the planet is heating up, the species migration from areas where there is not only malaria but also different viruses is simpler. How do we deal with these diseases if we don’t fully understand the structure of the mosquito genome? After all, we can’t and don’t want to just kill the whole species – it’s part of food chains.
Your plans also include joint work with the Mariinskaya Hospital. What will it involve?
We’ve started a large project on cardiology. We’re actively working with the Mariinskaya Hospital – we’re conducting the analysis of genome and phenotypic data. We’re trying to establish what combinations of genes and phenotypes are connected with a heightened risk of different heart diseases.
Now the situation is such that for most diseases, we know of the related correlations with some genes, genomic pairs, and so on. However, there’s only so much value to correlation – it’s possible for the amount of ice cream sold to correlate with the number of shark attacks. We want to get away from this and move on to explaining how this works.
As of now, there is a division between the sequences we find in the DNA and the annotation. We want to merge them into one structure. We want a result where we can poke at any part of a genome and say, this place is responsible for that thing. Currently, the amount of these places is insultingly low even in the human genome, not to speak of the others.
(author Konstantin Krylov)